Секвенирование — это метод, который используется для исследования ДНК и РНК и входящих в них белков. Первый геном человека был секвенирован в 2001 году. На это ушло более 10 лет исследований и около 3 млрд долларов. А сегодня секвенирование генома — достаточно рядовой анализ, хотя и не самый дешёвый: его стоимость составляет около 1000 долларов. Миллионы образцов человеческих геномов уже отсеквенированы.
Зачем нужно секвенирование генома?
Одна из важнейших целей секвенирования — уточнить диагноз у людей, которые страдают от генетических заболеваний. Но заметная доля известных геномов — это геномы условно здоровых людей. И здесь возникает вопрос — а стоит ли тратить миллионы долларов на «ковровое» секвенирование здоровых людей, неужели нельзя потратить эти деньги как-то более разумно?
Геном человека содержит около 6 млрд пар нуклеотидов. Но только в небольшой части генома закодирована информация о строении белков. Эта часть генома называется экзом.
Примерно 4–5 млн нуклеотидов [1] — это индивидуальные варианты, которые находятся в областях, кодирующих белки, или в непосредственной близости от них. Благодаря этим участкам генома мы отличаемся друг от друга. Если мы изучаем геном больного человека, то именно среди них нужно искать варианты, которые могли стать причиной заболевания — так называемые каузативные варианты.
Небольшое отступление: в генетике сейчас не принято использовать слово «мутация». Это считается неэтичным. Вместо этого используют словосочетание «каузативный вариант» (от слова cause — «причина»).
Как изучают генетические заболевания
Чтобы изучить генетическое заболевание, сначала нужно найти все индивидуальные варианты. Для этого сравниваются полученные данные с референсным геномом, избавившись от «ненастоящих» вариантов, возникших из-за ошибок секвенирования. Этим занимаются биоинформатики. Затем клинические интерпретаторы производят поиск «того самого» варианта, который может быть причиной заболевания.
Генетические заболевания глобально делятся на моногенные и полигенные. Моногенные вызваны патогенным вариантом в одном гене, а полигенные — множеством патогенных вариантов в разных генах. Конкретно в нашей статье мы говорим про поиск каузативных вариантов при моногенных заболеваниях.
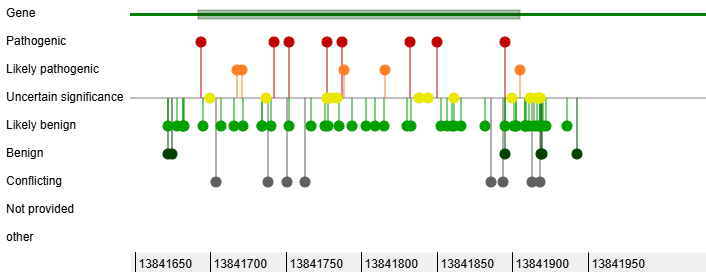
Позиции известных патогенных (красные и оранжевые) и безвредных (светло- и тёмно-зелёные) вариантов в гене DNAH5. DNAH5 кодирует часть аксонемного динеина — белка-моторчика, который приводит в движение реснички на поверхности мерцательного эпителия (в этом видео можно посмотреть на визуализацию работы динеина в жгутике). Скриншот из базы данных ClinVar [3]
Известные патогенные варианты можно выявить, сверившись со специализированными базами данных — PubMed [2], ClinVar [3]. Но что, если в геноме не оказалось ни одного известного варианта, который бы объяснял заболевание? Как среди упомянутых 4–5 млн вариантов найти каузативный? Тут-то интерпретатору и понадобятся знания о том, как выглядят геномы здоровых людей. Один из главных параметров, на который нужно обращать внимание, когда мы ищем причину заболевания, — это как часто встречается вариант. Поэтому первое, что делает интерпретатор, — отфильтровывает распространённые в популяции варианты.
Поиск патогенных вариантов
Патогенные варианты крайне редко встречаются у здоровых людей. Например, для возникновения аутосомно-доминантных заболеваний хватает мутации в одной копии гена. В случае тяжёлых заболеваний шансы встретить такой вариант в выборке геномов здоровых людей близки к нулю.
То есть первое, что делает интерпретатор, — смотрит на крайне редкие варианты. То есть такие, которые либо вообще не встречаются в выборках здоровых людей, либо встретились там максимум несколько раз в случае, если заболевание не очень тяжёлое.
Далее нужно понять, а могут ли выбранные варианты являться причиной заболевания.
Обычно в таких случаях делают секвенирование по Сэнгеру у родителей пациента — секвенируется короткий участок конкретного гена. Если вариант унаследован от здорового родителя в случае тяжёлого аутосомно-доминантного заболевания, то из этого следует, что вариант безвреден.
Базы данных с геномами здоровых людей
И тут мы вплотную подходим к вопросу, зачем нам нужны базы данных геномов здоровых людей. Если бы изначально у интерпретатора была база данных с вариантами конкретной популяции, то он бы сразу увидел, что вариант встречается у здоровых людей и безвреден. Это значит, что необходимость проводить проверку этого варианта и дополнительные дорогостоящие исследования не возникла бы. Не говоря о том, что не всегда оба родителя пациента бывают доступны.
В случае рецессивных заболеваний алгоритм слегка другой, но логика схожа. Ребёнок в таком случае наследует два патогенных варианта — по одному от каждого из родителей. Нужно проверить, встречались ли выбранные патогенные варианты в базах данных здоровых людей в гомозиготном состоянии (то есть в двух одинаковых аллелях). Если да, то вариант, скорее всего, безвреден.
Существует множество баз данных, которые помогают при поиске неизвестных прежде каузативных вариантов. Одна из таких баз — GnomAD [4]. Эта база, которая содержит информацию о вариантах, полученных от секвенирования условно здоровых людей. GnomAD — это самая крупная бесплатная база, в ней содержится 76 215 геномов и 730 947 экзомов.
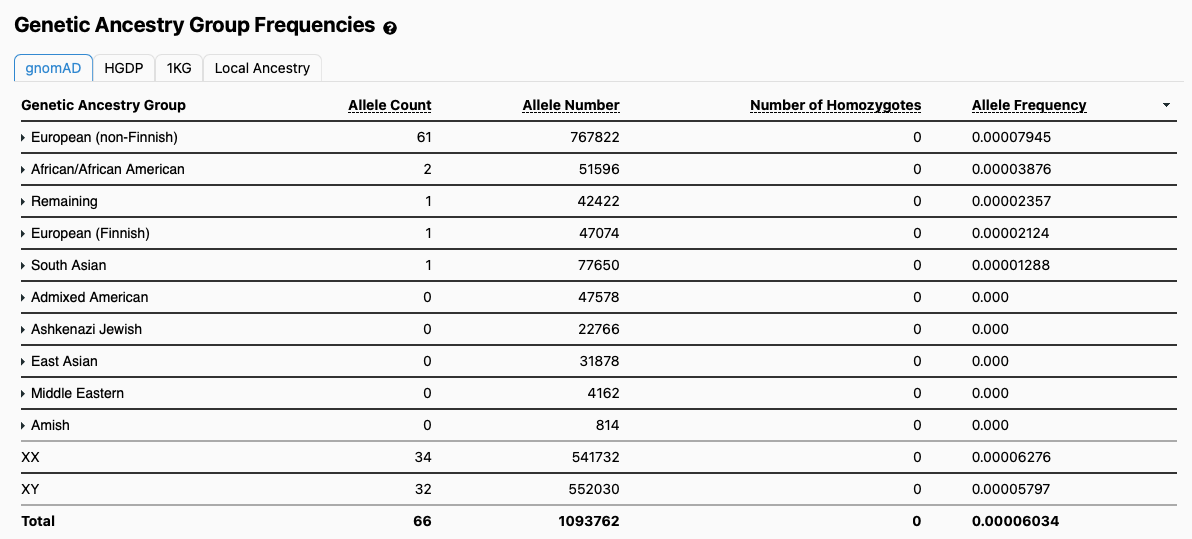
Скриншот из базы данных GnomAD [4]
В качестве примера (см. скриншот из источника) рассмотрим патогенный вариант chr5:13841895 G-A (это значит, что на пятой хромосоме в позиции 13841895 вместо нуклеотида G стоит нуклеотид A) в гене DNAH5. Allele count говорит, сколько раз именно этот вариант встретился в популяции, а allele number — сколько всего раз эта позиция была отсеквенирована в этой популяции. В гомозиготном состоянии такой вариант может привести к развитию цилиарной дискинезии — нарушению работы ресничек, приводящему к хроническим заболеваниям дыхательной системы. Поэтому закономерно, что в популяциях здоровых людей этот вариант в гомозиготном состоянии не встречается.
Нам нужно больше геномов
Но геномов и экзомов нужно ещё больше! Почему? Большая часть данных получена от европейцев, далее с большим отрывом идут американцы. И в этом тоже есть некоторая проблема. На протяжении всей истории человечества разные популяции проходили через бутылочные горлышки, и благодаря этому у многих малых народов образовался пул своих собственных редких вариантов, как доброкачественных, так и патогенных.
В базе GnomAD отдельно вынесены популяции финнов, амишей и евреев Ашкенази. Но этого явно недостаточно, и огромное количество популяций в мире всё ещё остается непредставленными. Например, в GnomAD в отдельную группу вообще не выделены популяции Африки, они входят в группу Remaining — остальные. А ведь африканская популяция — это самая генетически разнородная группа.
А что в России?
Для России эта проблема тоже весьма актуальна. У нас очень много разных малых народностей, и очевидно, что геном жителя Кавказа будет отличаться от генома жителя Якутии. Поэтому нам остро необходима собственная база данных, которая бы учитывала генетическое разнообразие народов нашей страны.
И в октябре 2024 года она у нас появилась — «База данных популяционных частот генетических вариантов населения Российской Федерации» [6]. И хотя российская популяция там не поделена на более мелкие популяции, представленная в базе информация бесценна для генетиков и клинических интерпретаторов: она помогает отсекать распространенные в российской популяции варианты и более точно находить причину заболеваний.
Источники
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015). https://doi.org/10.1038/nature15393.
- PubMed. [Электронный ресурс] URL: https://pubmed.ncbi.nlm.nih.gov/ (дата обращения: 03.11.2024).
- ClinVar. [Электронный ресурс] URL: https://www.ncbi.nlm.nih.gov/clinvar/ (дата обращения: 03.11.2024).
- GnomAD. [Электронный ресурс] URL: https://gnomad.broadinstitute.org/ (дата обращения: 03.11.2024).
- База данных популяционных частот генетических вариантов населения Российской Федерации. [Электронный ресурс] URL: https://nir.cspfmba.ru/info (дата обращения: 08.11.2024).